
CURRENT ISSUE
EDITOR'S PICKS
Review Article
How we manage a high D-dimer
ARTICLES IN THREE SENTENCES
Letter
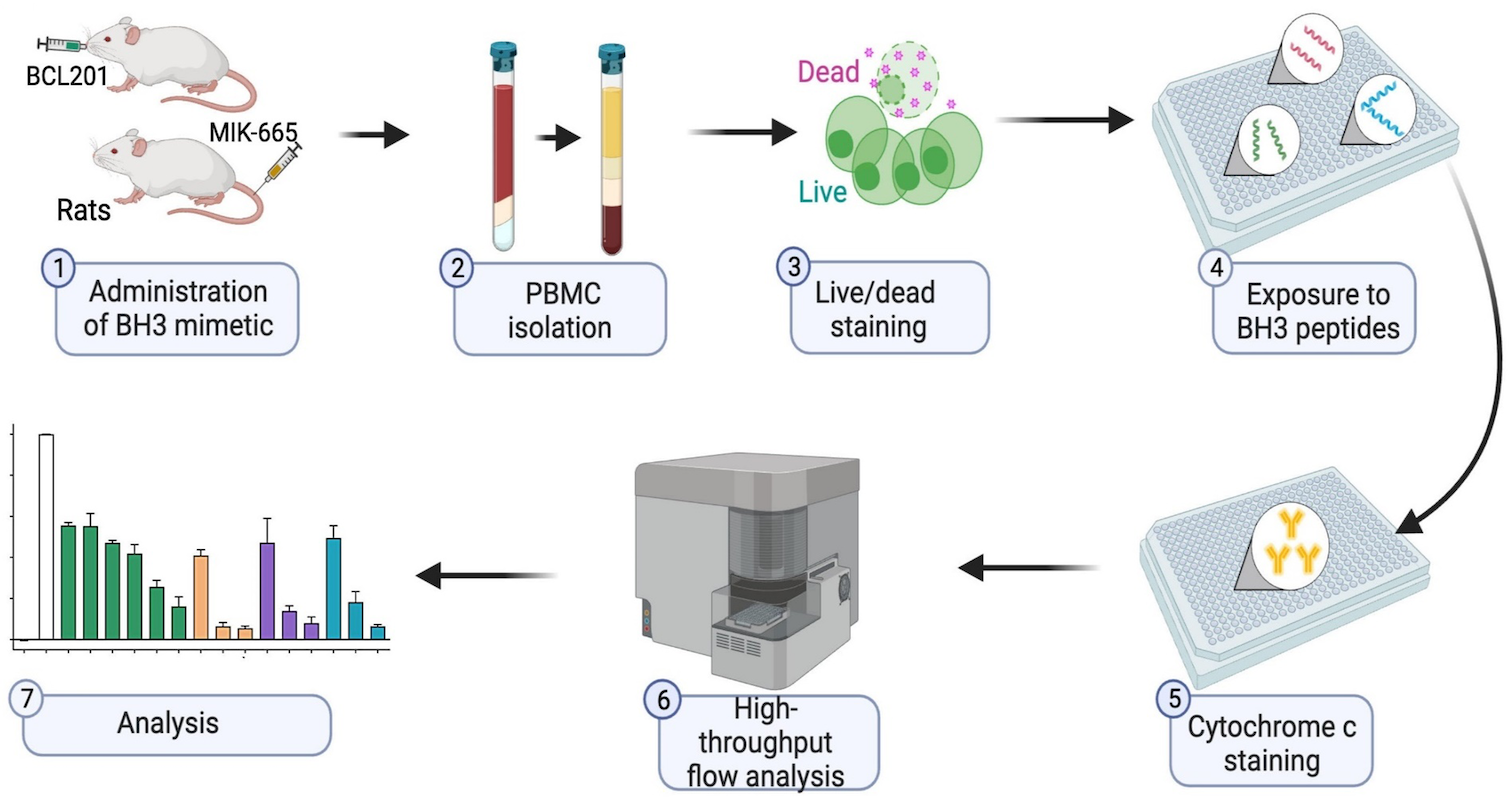
BH3 profiling as pharmacodynamic biomarker for the activity of BH3 mimetics
After the approval of the BCL-2 inhibitor venetoclax, the development of BH3 mimetic drugs that target anti-apoptotic BCL-2, BCL-XL, and MCL-1 proteins has been accelerated. For the successful development of candidate drugs, the availability of pharmacodynamic (PD) biomarkers would be important. Pan and colleagues demonstrated that the BH3 profiling, which determines the different ways a cell depends on BCL-2 anti-apoptotic proteins, is a robust and convenient peripheral blood-based PD biomarker for the activity of BH3 mimetics.
Article
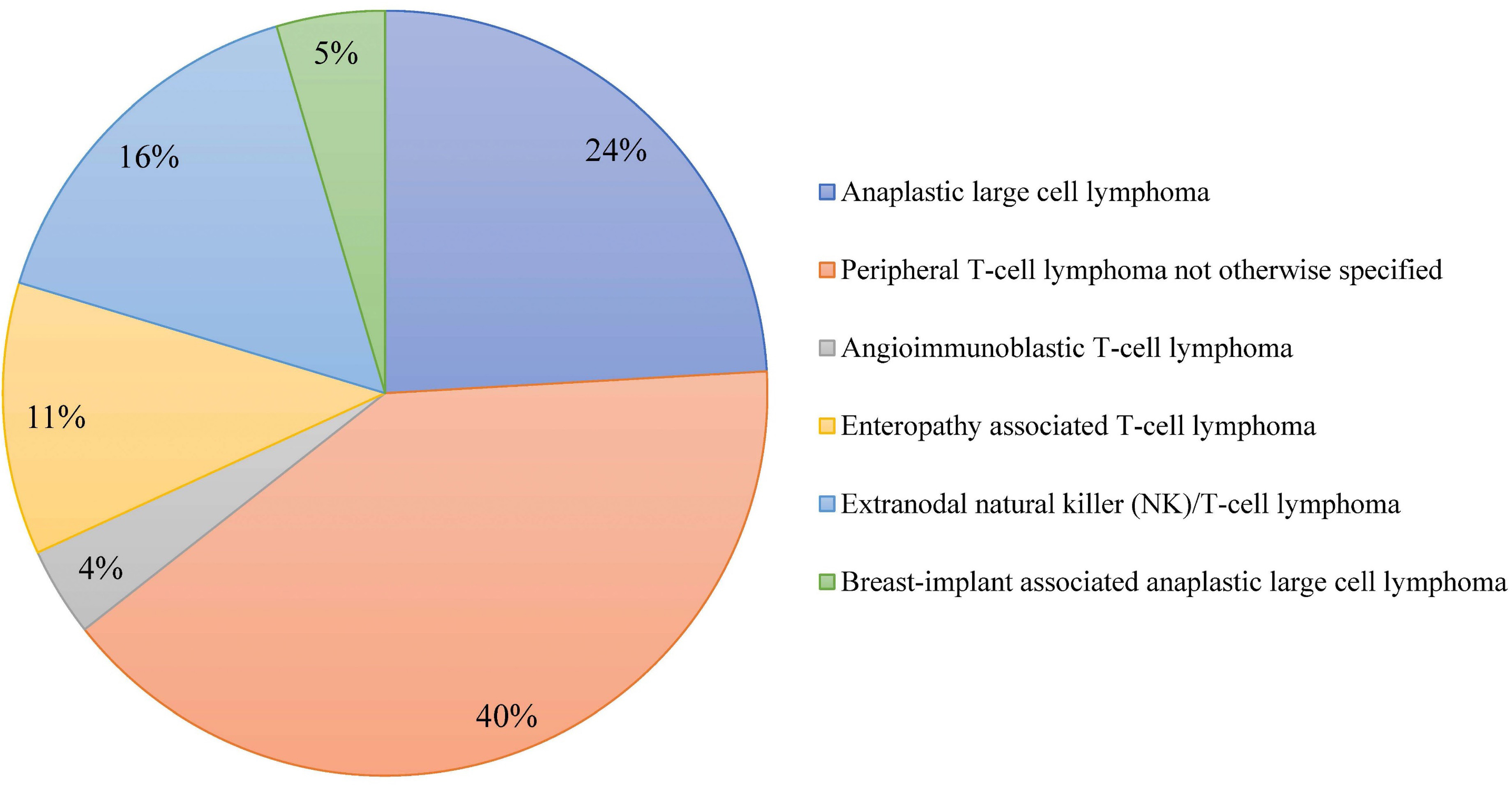
Outcome of combined modality treatment in first-line for stage I(E) peripheral T-cell lymphoma; a nationwide population-based cohort study from the Netherlands
A minority of patients with Peripheral T-cell lymphomas (PTCL) present with limited-stage disease and the optimal treatment for this subgroup is unknown. The aim of the nationwide population-based cohort study conducted by Meeuwes and colleagues is to describe first-line treatment and outcome of patients with stage I(E) PTCL identified between 1989 and 2020 in the Netherlands Cancer Registry. They found that for stage I(E) PTCL, the 5-year OS is 59% and a superior outcome is observed in patients treated with chemotherapy combined with radiotherapy.
Article
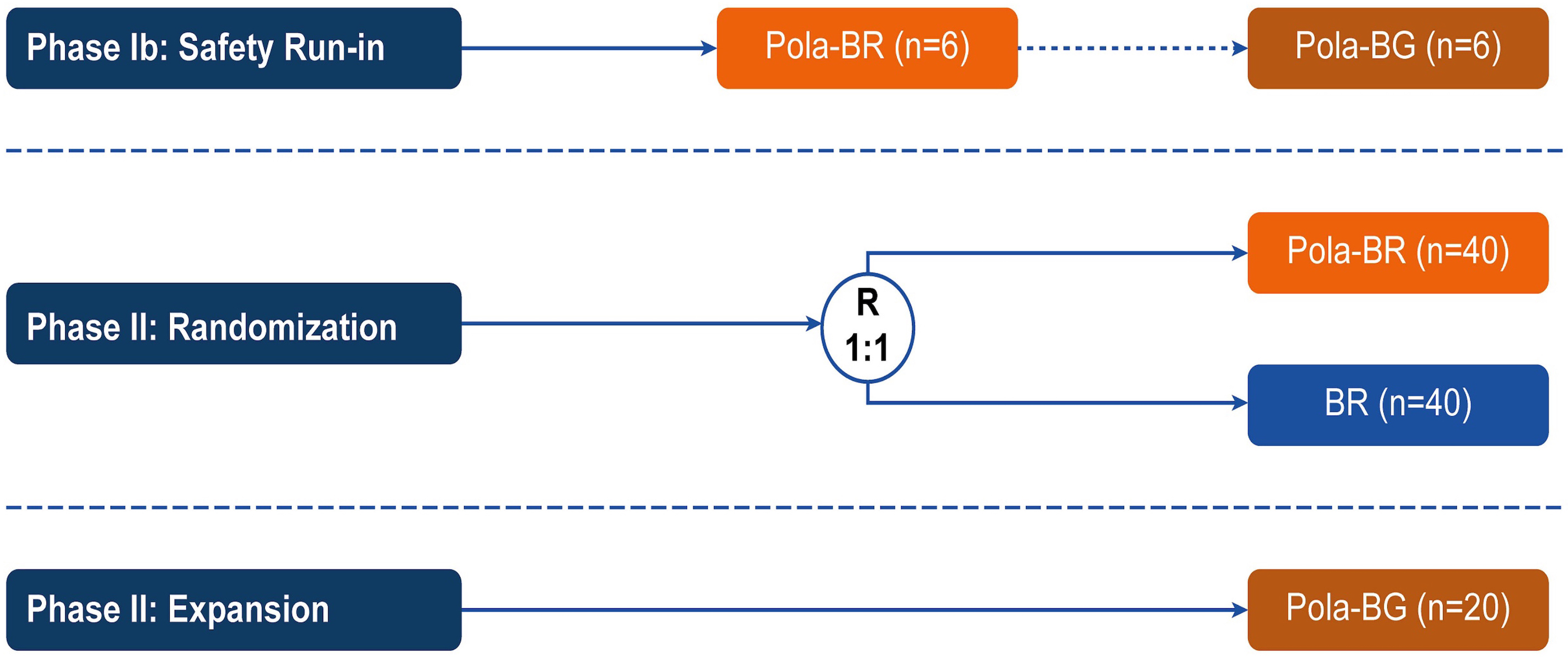
Polatuzumab vedotin plus bendamustine and rituximab or obinutuzumab in relapsed/refractory follicular lymphoma: a phase Ib/II study
Patients with relapsed/refractory (R/R) follicular lymphoma (FL) require new treatment options. In the phase Ib/II GO29365 study, the safety and efficacy of polatuzumab vedotin plus bendamustine and rituximab (Pola-BR) versus bendamustine and rituximab (BR) alone, and polatuzumab vedotin plus bendamustine and obinutuzumab (Pola-BG) were evaluated in patients with R/R FL. Flowers and colleagues present efficacy and safety results of the trial that do not demonstrate a benefit of adding Pola to BR or BG regimens for patients with R/R FL.
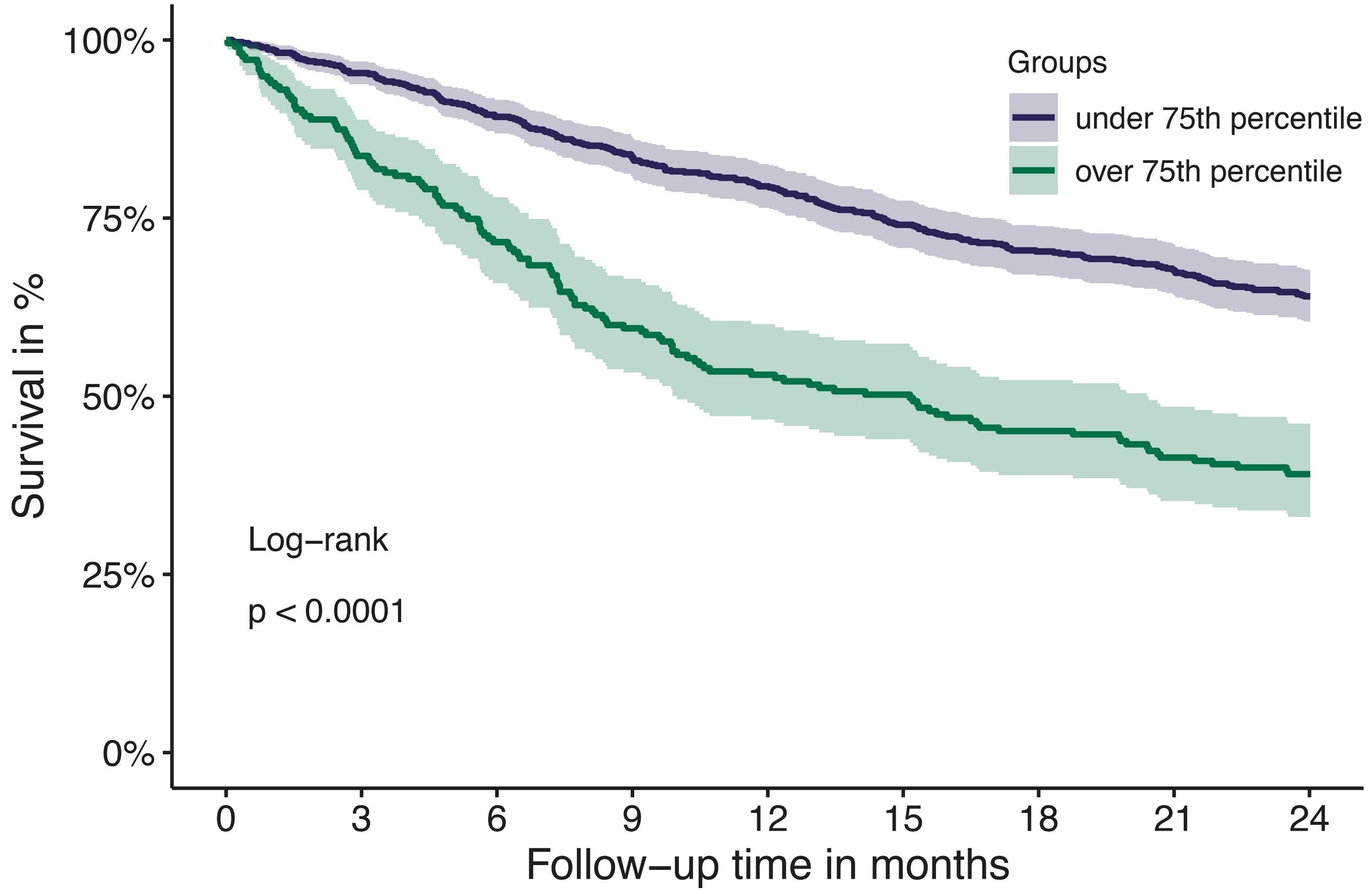
Article
Tissue factor pathway inhibitor is associated with risk of venous thromboembolism and all-cause mortality in patients with cancer
The risk for venous thromboembolism (VTE) in patients with cancer is about 7-9-fold higher compared to the general population. The mechanisms of cancer-associated thrombosis are not completely understood and its correlation with natural inhibitors of hemostasis is unclear. Englisch and colleagues investigated the association of the tissue factor pathway inhibitor (TFPI), a natural anticoagulant, with risk of VTE in patients with cancer, and found that increased TFPI levels are associated with VTE risk and with all-cause mortality.
TAKE ADVANTAGE FROM HAEMATOLOGICA



